SYNOPSIS
OPTIONS
DESCRIPTION
Edit distance (Damerau-Levenshtein distance)Edit distance measures how many insertions, deletions and substitutions are needed in order to convert one sequence into another. Suppose that you want to convert the letters in "DESK" into the letters in "RAVEN". The following figure shows all of the possible insertions, deletions and substitutions which can be done to their letters when converting between the two words.
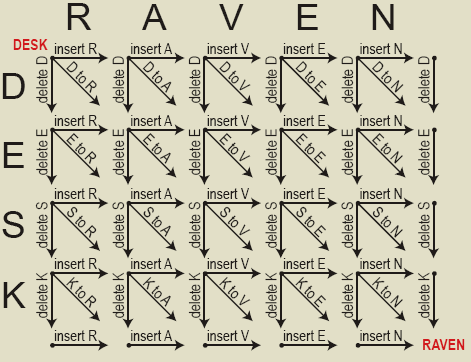
There are many paths through the above grid which can be followed from "DESK" at the top left corner to "RAVEN" in the bottom right corner. However, the most interesting path(s) is the one which takes the fewest steps. In this case, it takes at least five steps to get from DESK to RAVEN. The following figure highlights four possible paths which each require five edit operations. Substituting the letter "E" in DESK for the letter "E" in RAVEN is assigned zero cost, while all other segments are given a cost of one unit.
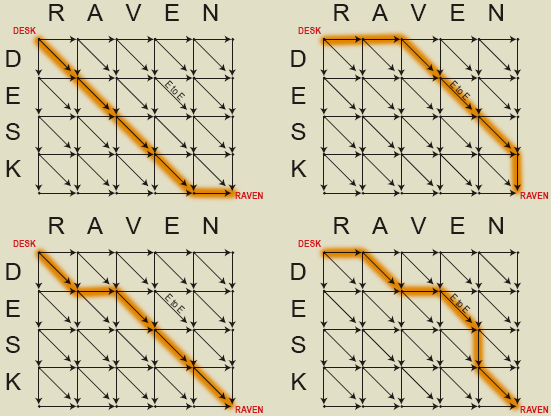
In each case, the paths require five changes to go from DESK to RAVEN. No path through the set of edits can have fewer than five steps, and the maximum value of nine is equivalent to following along the edges in either direction. Here are the intermediate text states for the each of the above paths:
Basic simil usageThe simil program requires two inputs: (1) a source file in the Humdrum file format, and (2) a search template file containing only a sequence of search strings. The resulting output is a Humdrum file containing a list of similarity measurements.
simil source template > output
In this case, each line of the output file gives the similarity of the template sequence to a sequence in the source starting on the same line of data. For example, the first similarity value of 0.51 is the similarity value between the string "ABC" in the template when compared to the first three data tokens in the source file: "XAB". A similarity value of 1.00 indicates an exact match, so notice that the second similarity value compares "ABC" in the template to "ABC" in the source file starting on the second line of data. Values towards zero indicate less similarity between the search template and the source data. By default, simil scales the edit distance scores by the equation: e-d/L, where d is the raw edit distance score, L is the number of elements in the template sequence, and e is 2.718281828.... If you want to see the raw edit distance scores which are inverted such that 0 indicates an exact match and larger values indicate less similarity, then use the -R option. The -n option is a unnormalized variant of the default output, where the similarity score is mapped with the equation e-d. simil source template > output
The -R option makes is clearer what has happened in the comparison of "ABC" to "XAB". In this case the 2 is generated by deleting the "X" at the start of the source sequence, and adding a "C" at the end of the source sequence. Note that the search template elements and the source data tokens do not have to be single-letter values, but can be any arbitrary strings. The simil command treats the entire string as a single unit when comparing elements between the target and source data. Note that it does not analyze internal similarities between strings, so "cat" and "hat" may seem similar, but as string elements being compared, simil will determine that they are not equivalent in any way. Here is an example of using simil with longer source and template tokens: simil source template > output
In this case, the highest similarity (0.56) occurs when the sequence "ominously, phantasia, and phantasma" lines up between the source and the search template. The value 0.37 represents the case when there is no similarity between the source and template. Editing weightsBy default, all editing operations cost the same amount (1.0 units). However, you can alter the default values in two ways: (1) by loading them from a initialization file, or (2) by changing the weights from the command-line. A file containing the new editing weights can be read by using the -w option followed by the name of a file which contains the weights. The weights file can contains lines of comments which start with #, and lines of code which start with the edit rule name followed by the revised score for that rule. Here is a sample file (not all rules have to be given in the file, only the ones which differ from the default weights of 1.0):
Alternatively, individual editing weights can be set from the command line as in the following example:
simil --s0 1.6 --s1 0.7 --s3 0.7 source template > output
By specifying the -W option, a list of editing weights will be appended to the output data as global comments:
simil -W --s0 1.6 --s1 0.7 --s3 0.7 source template > output
Sub-sequence template matchingBy using the -x option, you can search for the best substring match from the template in the source at each point. For example, with a template of the letters "ABC" and the option -x 2, all substrings of length two which can be generated from "ABC" will be compared to the source data, and a list of the top matching subsequences will be included in the output analysis. simil -x 2 source template > output
The **simixrf contains a list of integers which indicates the position of the substring(s) in the template which best match the source file at each position. In this case there are two substrings being compared to the source data: (1) "AB" and (2) "BC". When there is a similarity measurement tie between two or more substrings, indexes for all best-matching substrings are displayed, separated by commas. Various optionsTo output an extra spine containing the tokens sequence used from the source file for a particular simil score, use the -s option. Additionally, the -S (capital S) option can be used to suppress a space from being printed between the successive elements in the sequence. simil [-s [-S]] source template > output
To selectivly print information about only higher similarity values, use the -t option to suppress printing of data of low-similarity. For example, adding -t 0.70 to the command line options will prevent any similarity value less than 0.70 from being displayed in the output: simil -Sst 0.7 source template > output
In the output, lines with simil scores less than the threshold value are displayed as null tokens so that the relative locations of the values are preserved. You can remove the null token lines by piping the output into the Humdrum command "rid -d". EXAMPLES
Beyond knowing that the GFGAGAGFGAGAGFF sequence occurs 270 times in the sources, it may be useful to know where these sequences occur in the music. To do this, the simil command can be used to search the pitch sequence of the music and the value of 1.00 can be searched for in the resulting analysis spine. Here is the analysis for Exultet from the manuscript Av:
Scroll through the output column above and look for a value of 1.00 in the first column. Whenever this occurs, the sequence GFGAGAGFGAGAGFF will be found in the second column starting on that line. To see patterns in the occurrence of the sequence, it is more illuminating to plot the edit-distance similarity measurements throughout the music as shown below.
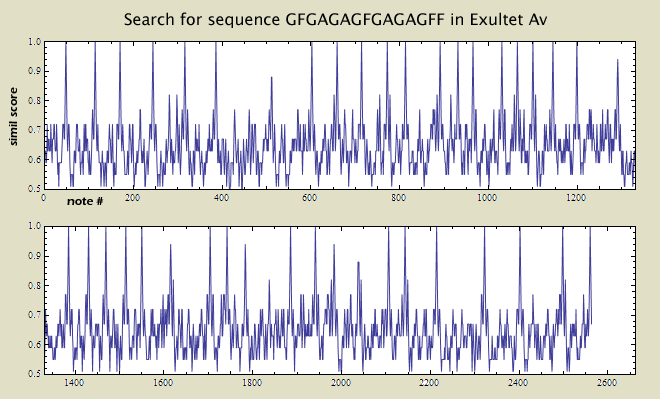
In Exultet source M2, however, the sequence GFGAGAGFGAGAGFF never occurs. Instead, the sequence has been altered. simil source template -p > output
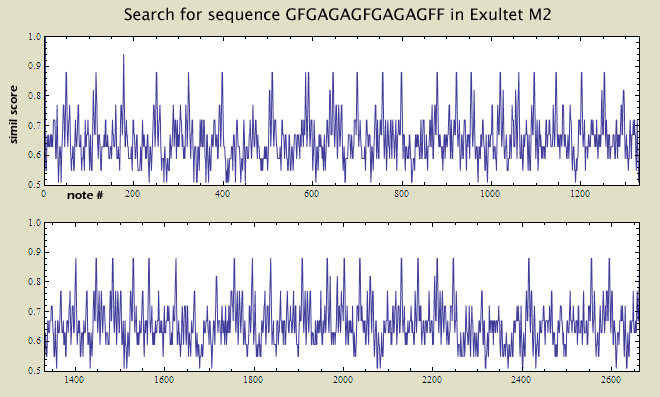
simil source template -Ss -t 0.8| \ grep ' GF' | cut -f2 | sort | uniq -c | sort -nr
Av: GFGAGAGFGAGAGFF
M2: GFGAGAGF AGAGFFG
When comparing the two sequences, the last G in M2 should be ignored
since the limit of 15 notes in the analysis may have chopped the G from
the end of the sequences in the Av source.
SEE ALSODOWNLOAD
The source code for the program was last modified on 8 Dec 2009. Click here to go to the full source-code download page. |
